

V minulom blogu som vám vysvetlila, ako fungujú prístroje na sekvenovanie DNA. V súčasnosti už veda pokročila tak, že sekvencie genómu nejakej novoobjavenej baktérie môžeme získať do 48 hodín. Rýchlosť a efektivita je veľmi dôležitá hlavne vtedy, ak sa z ničoho-nič objaví nejaká nebezpečná baktéria, ktorá zabíja jedného človeka za druhým. Počítam, že sekvencie by sa dali získať do 48 hodín, ak túto novú baktériu dostane do rúk schopná laboratórna technička, ktorá za jeden deň zvládne extrahovať DNA, pripraviť DNA to takej formy, aby ju vedel prečítať sekvenátor (volá sa to pripraviť knižnicu - library preparation) a cez noc nechá bežať PCR reakciu, v ktorej sa namnožia kúsky DNA na guličkách v takej olejovej kvapke (o tejto technike som písala v minulom blogu). Na druhý deň ráno rýchlo pripraví guličky s kúskami DNA do takej formy, aby sa mohli naniesť do sekvenátora a večer spustí sekvenátor, aby bežal celú noc. Na ďalší deň ráno nám z neho vylezú sekvencie. Čo ale teraz s nimi?
Teraz nasleduje práca pre šikovného bioinformatika. Väčšina šikovných bioinformatikov trávi veľkú časť svojho času programovaním, pretože sa snažia vytvoriť nejaký skript, kde bude možné jediným stlačením tlačítka Enter urobiť všetko spracovanie dát automaticky a rýchlo. To je veľmi dôležité, pretože sekvenačné prístroje pracujú v súčasnej dobe naozaj rýchlo, ale dostávame sa do obmedzení so spracovávaním dát v počítačoch, ktoré musí byť urobené správne a tiež rýchlo. Mnohé z pokusov, ktoré robíme, sú jedinečné, priekopnícke a nerobilo ich predtým veľa ľudí, takže v takýchto situáciách nie je možné používať žiaden program s pekným užívateľským prostredím s ikonkami, na ktoré sa kliká myšou.
Keby sme mali času neúrekom a tiež aj veľmi pevné nervy, úplne teoreticky by sme mohli zložiť sekvencie genómu nejakého organizmu aj ručne. Ale je to skoro nemožné, veď si to len predstavte! Počas jedného sekvenovania naraz dostaneme napríklad 100 000 sekvencií o dĺžke 600 nukleotidov. Musíme ich zložiť, aby sme dostali genóm nejakej baktérie o predpokladanej dĺžke 4 000 000 nukleotidov. Prečítame si prvú a začneme ju porovnávať so sekvenciou číslo 2, 3, 4... a skúšame, či niektorá z nich bude mať začiatok taký istý ako mala prvá sekvencia koniec. To by nám teda trvalo strašne dlho! Vďakabohu máme na to počítače a hlavne šikovných bioinformatikov.
Nie je to ale také jednoduché, pretože je tu pár problémov s chybičkami v sekvenciách a rôznych otázok v metodike priraďovania sekvencií. Je zámena alebo zmiznutie nejakého písmenka naozajstná mutácia alebo je to len nejaká chyba v sekvenovaní? Ak je začiatok druhej sekvencie zhodný s koncom prvej sekvencie s výnimkou tohoto jediného písmenka, má sa druhá sekvencia priradiť k prvej alebo má sa nechať bokom? Koľkokrát sa musí chybička vyskytnúť, aby sme potvrdili, že je to naozaj mutácia a nie chyba sekvenovania? Čo ak sú prvé polovice sekvencií číslo 1 a číslo 2 rovnaké, ale zbytok je rôzny? Majú sa takéto sekvencie priradiť k sebe alebo nechať oddelene? Čo urobíme so sekvenciou, ktoré je krátka a mohla by sa priradiť k niekoľkým rôznym skevenciám? Podľa čoho sa rozhodneme, ku ktorej sekvencii ju priradíme? Alebo necháme ju radšej bokom? Odpovede na tieto otázky budú iné v prípade sekvenovania genómu jedného jediného organizmu a iné v prípade sekvenovania mnohých organizmov naraz - metagenomike (o tom viac v predchádzajúcom blogu). A špeciálny prístup bude potrebný, ak sekvenovenujeme cDNA (takže vlastne len gény, ktoré sa prepíšu v danom momente do RNA) alebo na identifikovanie baktérií pomocou špeciálneho identifikačného génu. A poskladať niekoľkomilionové genómy veľkých organizmov je výzvou aj pre tie najvýkonnejšie počítače.
Samozrejme, najrýchlejšie je skladanie nejakej novej baktérie, ktorá sa podobá na nejakú už predtým osekvenovanú baktériu. V takomto prípade môžeme tú predtým osekvenovanú baktériu použiť ako vzor, podľa ktorej priraďujeme a zlepujeme nové sekvencie. Ak zistíme nejaký rozdiel, snažíme sa zistiť, odkiaľ tieto nové sekvencie prišli. Možno sa len nejako premiestnili v rámci genómu toho istého druhu baktérie alebo sa do genómu dostali nejaké úplne nové gény. Takto sa napríklad zistilo, že mediálne známa smrtiaca baktéria z Nemecka z leta roku 2011 je vlastne E.coli, ktorá prevzala nebezpečné gény od Shigelly, čo sa volá horizontálny transfer génov (o tom niekedy nabudúce).
Všetky tieto prístupy majú jedno spoločné: sekvencie, ktoré dostaneme sa priprovnávajú už k predtým zistením sekvenciám, ktoré sú uložené v databázach na internete. Tu je tiež otázka, aké nastavenia použijeme na definovanie zhodnosti sekvencii? Hlavne musíme to vedieť robiť automaticky, lebo ručne identifikovať niekoľko miliónov sekvencií by bolo časovo veľmi neefektívne. Aby som vám ukázala, že to nie je také jednoduché, môžete si teraz sami skúsiť identifikovať jednu moju bakteriálnu sekvenciu. Skopírujte si nasledujúce písmenká (je tu 635 písmen):
gagcggccgccagatcttccggatggctcgagtttttcaagaagattcgtgactcatccc
gcggcggctggcacggagttagccgatccttattcgtagggtacatacaaagcggaacac
gttccgcactttattcccctataaaagaagtttacgacccatagggccttcatccttcac
gctacttggctggttcagactctcgtccattgaccaatattcctcactgctgcctcccgt
aggagtttggaccgtgtctcagttccaatgtgggggaccttcctctcagaacccctatcc
atcgaagacttggtgagccgttacctcaccaacaatctaatggaacgcatccccatcaaa
caccgaaattctttaataacaataagatgccttatcgctatactatcgggtattaatctt
tctttcgaaaggctatccccgtgtgaatggtaggttggatacgtgttactcacccgtgcg
ccggtcgtcagcggtattgctaccctgctacccctcgacttgcatgtgttaagcctgtag
ctagcgttcatcctgagccaggatcaaactctgatgagtcacgaatcttgctgaaaaact
cgagccatccggaagatctggcggccgctctccca
Choďte na stránku NCBI BLAST
Tam vložte do okienka tieto písmená.
Pri "Choose Search Set" zaškrtnite "Database: Others"
Vyberte "Nucleotide collection (nr/nt)"
Pri "Program Selection" zaškrtnite "Highly similar sequences (megablast)".
Úplne dolu na stránke zaškrtnite "Show results in a new window" a kliknite na tlacitko BLAST.
V novej záložke sa vám objaví niečo podobné ako na nasledujúcej fotke. Je to sekvencia a cervenou farbou zobrazené jej dobré pokrytie a presnosť identifikovania. Pod obrázkom sa vám ukáže zoznam všetkých druhov baktérií, na ktoré sa sekvencia podobá. A pod zoznamom je presne ukázané, ako sa priradili naše sekvencie k organizmu v databáze.
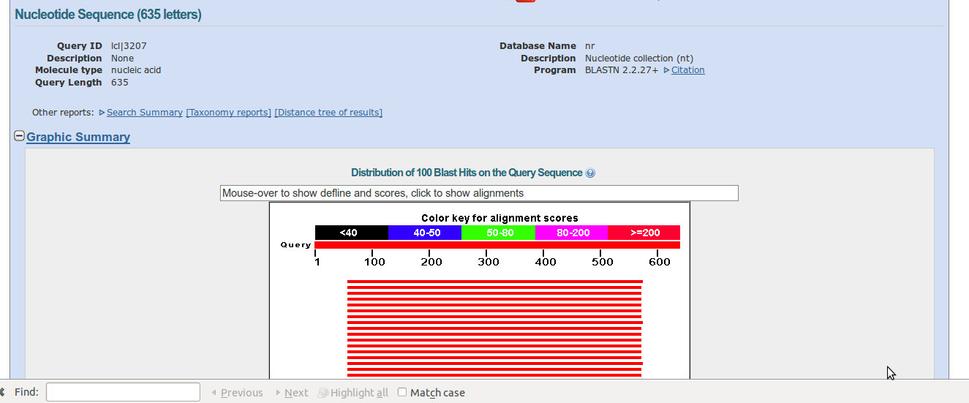
Je to baktéria Bacteroides. Ako vidíte, ukázalo sa, že naša sekvencia je pokrytá na 80%. Jej začiatok a koniec je nie pokrytý touto baktériou, lebo je to sekvencia zvyšku plasmidu, do ktorého bol tento kúsok DNA vložený, teda zaklonovaný (viac v minulom blogu).
Skúsme teraz vymazať nejaké prvé a posledné písmená, takže zostane len 420 písmen:
gttccgcactttattcccctataaaagaagtttacgacccatagggccttcatccttcac
gctacttggctggttcagactctcgtccattgaccaatattcctcactgctgcctcccgt
aggagtttggaccgtgtctcagttccaatgtgggggaccttcctctcagaacccctatcc
atcgaagacttggtgagccgttacctcaccaacaatctaatggaacgcatccccatcaaa
caccgaaattctttaataacaataagatgccttatcgctatactatcgggtattaatctt
tctttcgaaaggctatccccgtgtgaatggtaggttggatacgtgttactcacccgtgcg
ccggtcgtcagcggtattgctaccctgctacccctcgacttgcatgtgttaagcctgtag
Teraz už vidíte, že sekvencia je pokrýtá na 100% a s "Bacteroides sp. CannelCatfish9 16S ribosomal RNA gene" je zhodná na 99%. Jediný rozdiel je jediné písmenko, ktoré je v našom prípade C a v prípade baktérie Bacteroides je T.
Skúste teraz skopírovať prvý riadok z mojej originálnej sekvencie, teda:
gagcggccgccagatcttccggatggctcgagtttttcaagaagattcgtgactcatccc
Výjde vám veľa rôznych vecí, najlogickejší z nich je akýsi "Cloning vector". To je zbytok plasmidu, ktorý som použila na klonovanie a sekvenovanie. Okrem neho vo výsledku môžete vidieť aj veľa rôznych hlúpostí, medzi nimi nejaké bakterie a rastliny Dieffenbachia či pohanka. Domnievam sa, ze nejaki vedci neodstránili zo svojich rastlinných/bakterialnych sekvencií sekvencie plasmidov či sekvenačných adaptorov. Úlohou vedcov je vedieť robiť toto porovnávanie s databázami automaticky a hlavne správne a logicky.
A ak sa radi hráte, tak si vymyslite nejaké vlastné sekvencie DNA a skúste ich "blastovať" na uvedenej stránke podľa môjho vzoru. Pozor, aby neboli veľmi krátke, aby vám nevyšli nejaké hlúposti. Napíšte mi do diskusie, ako sa vám darilo. Dobre sa bavte!






