
Po roku 2000 sa začala rozvíjať technológia sekvenovania DNA od firmy Roche, ktorá dostala skratku 454. Už som spomínala na predchádzajúcich blogoch, že sekvenátory nevedia čítať celú molekulu DNA. Musí byť nakrájaná na kúsky. Na začiatku 454 sekvenátory vedeli čítať kúsky dlhé len pár desiatok písmen. Technológia sa stále zlepšovala a približne v roku 2011 už bolo možné sekvenovať kúsky dlhé až 1000 písmen.
Zároveň sa na trh začala predierať aj platforma Illumina. Najprv ju mnohí zatracovali, pretože v dobe, keď 454 už produkovalo aj 500 písmenkové sekvencie, sekvencie Illuminy boli dlhé len pár desiatok písmen. Okrem toho sekvenátor od Illuminy občas robil aj dosť chýb. Illumina sa ale časom tak zlepšila, že to dĺžkou sekvencií dotiahla až na 600 písmen (sekvenátor MiSeq). A najväčšou výhodou sa stalo to, že sa za 10 násobne nižšiu cenu dá získať 10 krát viac sekvencií. A to doviedlo 454 sekvenovanie k zániku, až firma Roche pred rokom oznámila, že 454 sekvenovanie v roku 2016 zanikne.
Vedci sa proste uchamtili na vysoký počet sekvencií a super cenu, ktorú Illumina ponúka. Úplne sa zadudlo na dĺžku. Dĺžka 600 písmen z MiSeq a 800 -1000 písmen zo 454 je však veľký rozdiel. Hlavne keď chcete identifikovať baktérie.
Už som tiež písala aj o tom, že baktérie majú jeden gén, ktorý sa volá 16S. Podľa neho vieme identifikovať akúkoľvek baktériu, ako keby to bol jej občiansky preukaz, je to asi 1500 písmen. Tento gén obsahuje variabilné regióny, kde sa jedno písmenko u jedného druhu baktérie zamení alebo zmizne u iného druhu. Tiež obsahuje aj zakonzervované regióny, kde sa skoro žiadne zámeny písmen neobjavujú. Sekvencie týchto zakonzervovaných regiónov sa dajú použiť na navrhnutie primerov, ktoré nám pomôžu dostať tento dlhý gén do takej formy, aby ho vedel sekvenátor prečítať. Robí sa to pomocou reakcie PCR.
O reakcii PCR som už písala tiež predtým. Využíva sa v nej vlastnosť enzýmu polymeráza, že dokáže k originálnej molekule DNA prikladať písmenká voľne plávajúce v skúmavke. Prikladá ich ale len na úseku vymedzenom dvoma primermi. Keď sa toto prikladanie písmenok zopakuje napríklad 30 krát, krátky úsek medzi primerami bude namnožený 1 073 741 824 krát (2 na 30-tu), zatiaľčo originálna DNA zostane len jedna jediná dlhá molekula, ktorú sekvenátor nedokáže prečítať.
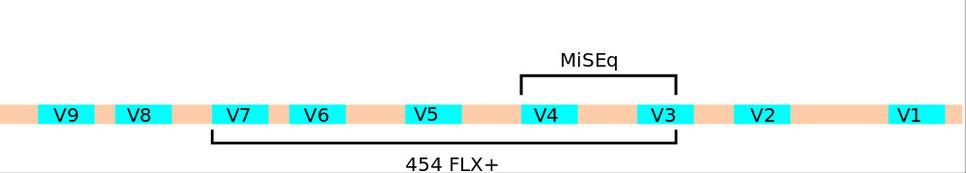
V praxi to znamená, že ak sekvenátor dokáže čítať len 600 písmen, musíme si z génu 16S vybrať na namnoženie len taký úsek, ktorý bude dlhý menej než 600 písmen. Na obrázku môžete vidieť, že variabilné regióny V3 a V4 sú dlhé tak akurát 550 písmen, čo sa do 600 písmenového obmedzenia sekvenátora MiSeq ešte vojde. S technológiou 454 FLX+, ktorá dokáže čítať až 1000 písmen, sa dajú osekvenovať okrem V3 a V4 ešte aj regióny V5, V6 a V7. To je veľmi potrebná informácia na identifikáciu baktérií.
Ja som nedávno začala spracovávť svoje prvé sekvencie zo sekvenátora MiSeq. Boli to regióny V3 a V4, dlhé 550 písmen. Moja radosť z 10 násobného množstva sekvencií za 10 krát menšiu cenu (v porovnaní s 454 FLX+) ma však prešla hneď, keď som sa pokúšala zistiť množstvo druhu Clostridium difficile v mojich fekálnych vzorkách. Ono to totiž síce funguje s inými druhmi baktérií, ale s Clostrídiom to nejde!
Rod Clostridium sa rozdeľuje na veľa druhov, niektoré z nich sú človeku prospešné, ale jeden z nich – Clostridium difficile je nebezpečný a dokáže dokonca spôsobiť aj smrť. Ja som chcela vedieť, či majú pacienti, ktorých skúmam, nejaké zvýšenie počtu Clostridium difficile vo svojich výkaloch.
Gén 16S má cca 1500 písmen a čím viac tých písmen osekvenujeme, tým precíznejšie dokážeme idektifikovať baktérie. Dokonca by sa dali rozpoznať aj bakteriálne kmene, ak by bola sekvencia dostatočne dlhá. Sekvenovaním regiónov V3 a V4 dokážeme rozpoznať niektoré druhy naozaj perfektne, ale o clostrídiu to neplatí - nedokážeme rozlíšiť ani Clostridium difficile od Clostridium perfringens! Výsledok, ktorý z tejto 550 písmenkovej sekvencie dostanem, bude označený ako „neznámy druh rodu Clostridium“. A teraz si mám domýšľať, či táto sekvencia patrí dobrému alebo zlému Clostrídiu?!
Dalo by sa to rozpoznať vytvorením takzvaného fylogenetické stromu. Tam sa všetky sekvencie potenciálne patriace C. difficile porovnajú a ak sa tam nájdu nejaké odlišnosti, tak sa to posunie do „vyššieho konára“ toho stromu. Podľa vzdialenosti „konárov“ by sa dalo určiť, ktorému druhu clostrídia sa moja sekvencia najviac podobá.
S takými krátkymi sekvenciami však nefunguje ani táto metóda, pretože napríklad Clostridium difficile a Clostridium perfringens sa v regiónoch V3 a V4 odlišujú len v 2-3 písmenkách. Ale tiež aj sekvenátor MiSeq občas robí nejaké chybičky. Sekvencie s takýmito chybičkami sa vo fylogenetickom strome zostavenom z tak krátkych sekvencií zdajú ako úplne nový druh clostrídia, ale to je nesprávne. Ak by som mala k dispozícii dlhšie sekvencie, týchto pár chybných písmeniek by bolo pre identifikáciu druhu úplne bezvýznamných a zostavil by sa mi krásny fylogenetický strom.

Pre ilustráciu som tu tak schématicky namaľovala taký vymyslený príklad, ako asi vyzerajú rozdiely medzi Clostridium difficile, Clostridium perfringens a Enterococcus faecium, čo je už, ako podľa mena vidíte, úplne iný rod. V géne 16S by sa rozdiely medzi nimi prejavily tak, že Enterococcus faecium tam bude mať jeden veľký kus sekvencie navyše, takže je jasné, že ani sekvenačná chyba by nedokázala spôsobiť nesprávne identifikovanie. Ako rozdiely medzi C. perfringens a C. difficile som si na obrázku vymyslela dve 4 zámeny písmen označené hviezdičkami. Ak sa do toho pripletie hoci len jednopísmenková sekvenačná chyba, nastane problém, že nebudem vedieť, aký druh clostrídia to je.
Čo mi teraz ostáva? 454 sekvenovanie už zaniká a aj keby nezaniklo, je pridrahé. Nuž zostáva mi len zmieriť sa s tým, že budem mať síce veľa lacných sekvencií, ale prítomnosť C. difficile u pacientov týmto spôsobom asi neodhalím. Na rozlúštenie tejto otázky existujú jednoduchšie metódy, ktoré mi presne určia množstvo C. difficile vo vzorke namnožením celej sekvencie génu 16S. Ale to nebude už sekvenovanie. Sekvenovaním môžem rozlúštiť zloženie baktérialnych rodov, môžem porovnávať aké rody baktérií sa premnožili v tráviacom trakte pacientov a aké sa nenachádzajú u zdravých jedincov. Ťažké je ale správne identifikovať druhy týchto rodov. Rôzne druhy, dokonca rôzne kmene baktérií môžu mať rôzne funkcie, jeden kmeň môže byť nebezpečný, ale druhý priateľský, a preto je ťažké tieto výsledky interpretovať.






